Biogen's Antibody for Alzheimer's Disease


Aducanumab Preclinical and Early Clinical Findings
In preclinical studies, Aducanumab was shown to bind amyloid plaque acutely, while chronic treatment helped clear beta-amyloid and restore calcium homeostasis in mice (Kastanenka et al. 2016). Findings from a phase 1b clinical study (NCT01677572) showed promising outcomes for the use of Aducanumab in patients with mild Alzheimer's disease. A once a month Aducanumab infusion for a full year led to a reduction in brain beta-amyloid plaques and slowing of clinical decline (Sevigny et al. 2016).
Phase 3 trials for Aducanumab were undertaken by Biogen in 2015 to assess efficacy and safety of two dosing regimens. By 2016, the FDA had granted fast track status for Aducanumab. However, results from a futility study in 2019, which indicated inability to meet end point objectives, resulted in early termination of the Phase 3 trials (studies 301 and 302) (Knopman et al. 2020).
Biogen Addresses Phase 3 Difficulties
Following a collaboration with the FDA to assess the findings of the interrupted Phase 3 trials, Biogen was granted a Biologics License Application (BLA) with Priority review for Aducanumab. Biogen's team discussed findings for studies 301 and 302 in their presentation to the FDAon November 6th, 2020, conceding that study 301 was a failed study due to an imbalance in target dose exposure and in the number of subjects with rapid progressive disease. However, Biogen's team argued that findings from study 302 supported the efficacy of Aducanumab: "These studies consistently demonstrate that sufficient exposure to 10 mg/kg Aducanumab is effective at reducing the clinical decline in patients with early symptomatic Alzheimer's disease".
The meeting's outcome was not encouraging, with the FDA's Peripheral and Central Nervous System Drugs Advisory Committee voting 8 against, 1 in support and 2 uncertain in relation to efficacy findings. Overall, the committee was not completely convinced that evidence from the positive 302 study strongly supported the effectiveness of Aducanumab for the treatment of Alzheimer's disease, as evidenced by 10 members voting against this view. Still the jury is out and the FDA has to make a final decision on drug approval by March 2021.
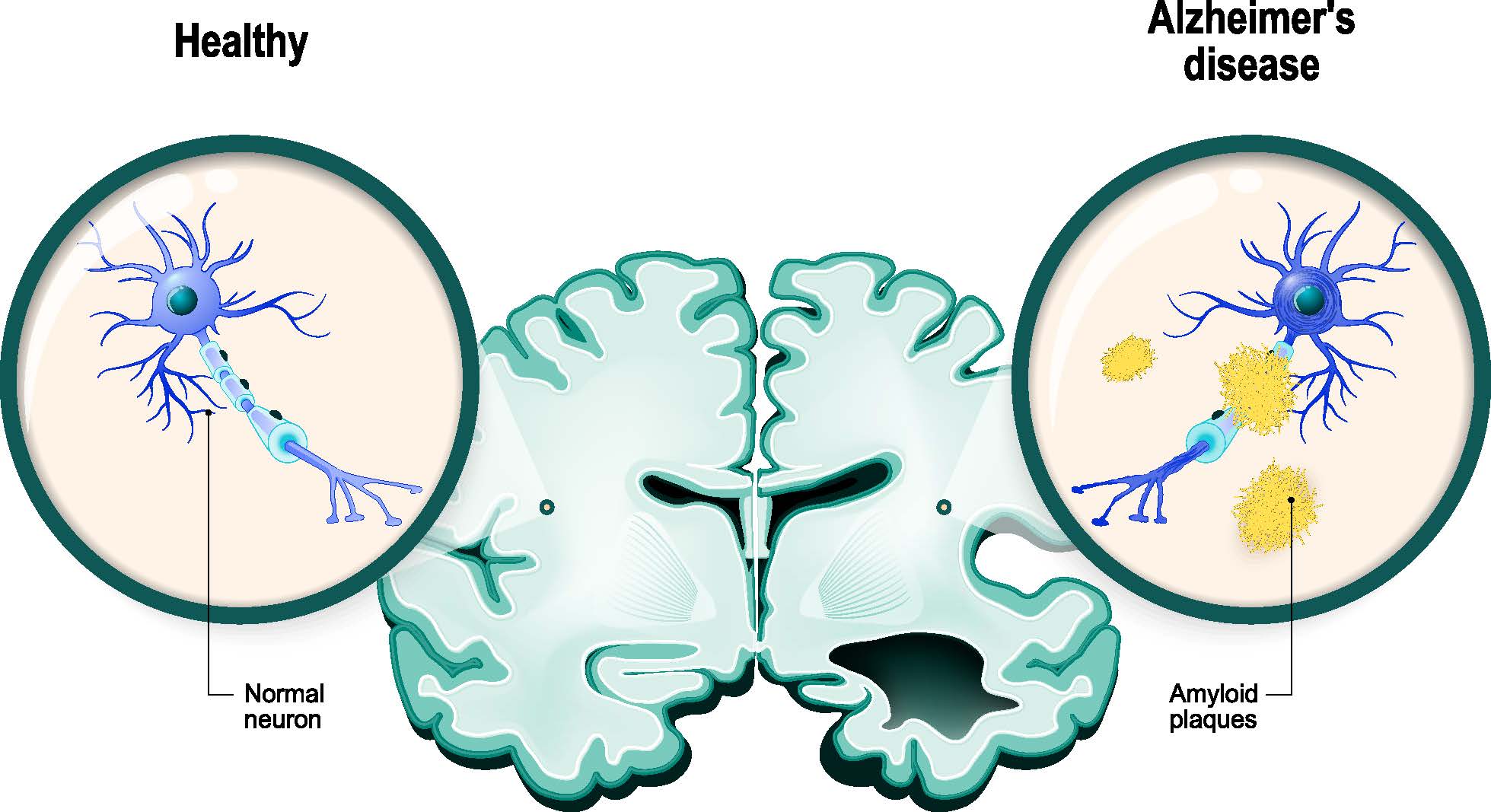
In Alzheimer's disease cognitive decline is proposed to occur as the result of beta-amyloid deposition. Amyloid aggregates, oligomers, fibrils and ultimately plaques, accumulate in the extracellular space further promoting pathology through the aggregation of tau. Dysfunction in neuronal function, inflammation and ultimately cell death follows with evident structural brain changes.
What is Alzheimer's Disease?
Alzheimer's disease belongs to a group of neurodegenerative diseases classified under the broad umbrella of proteinopathies. These diseases are characterized by the presence of aggregated proteins, which represents a common pathological hallmark. In Alzheimer's disease, several protein aggregates are associated with disease progression including intracellular beta-amyloid aggregates, extracellular beta-amyloid plaques, and intraneuronal tau-neurofibrillary tangles. Intracellular beta-amyloid aggregates are linked to degeneration and neuronal death, while extracellular beta-amyloid plaques lead to loss of microglia and senescence of stem and progenitor cells. Additionally, beta-amyloid plaques promote the formation of tau-neurofibrillary tangles, resulting in inflammation, oxidative stress, and microvascular pathology (Wang et al. 2016).
Alzheimer's disease progression is characterized by impaired cognition, which in the majority of cases is only evident at later life stages (> age 65). Currently, there is no cure for Alzheimer's disease and no therapies have been identified which may decrease progression or effectively ameliorate symptoms. Although the proposal that beta-amyloid aggregation and consequent tau accumulation are central to Alzheimer's disease is well accepted, other downstream mechanisms not fully elucidated appear to be at play. This is because beta-amyloid accumulation often fails to clearly correlate with the progressive cognitive decline observed in Alzheimer's disease (Long and Holtzman, 2019).
Reference
- Like (4)
- Reply
-
Share



About Us · User Accounts and Benefits · Privacy Policy · Management Center · FAQs
© 2026 MolecularCloud