Sanger Sequencing vs. Next-Generation Sequencing (NGS)

Evolution of DNA sequencing
DNA sequencing is the process of identifying the exact order of nucleotide bases (i.e., Adenine, Cytosine, Guanine, and Thymine) encoding specific genomic information. DNA sequencing technology rapidly advanced from its inception in the 1970s with the work of Frederick Sanger, who sequenced the first full genome based on the “plus and minus method” (aSanger et al. 1977). Ultimately, the Sanger chain termination or dideoxy method, also first reported in 1977, provided the foundation for fast-paced growth in DNA sequencing technologies and enabled sequencing the human genome for the first time in 2001 (bSanger et al. 1977).
Next-Generation sequencing (NGS) technologies evolved from the need to sequence larger volumes of genetic material faster and at a lower cost. Over the years, different NGS methods and platforms have been developed based on unique chemistries and detection methods (e.g., Pyrosequencing, Reversible-dye terminator, and Proton detection) (Garrido-Cardenas et al. 2017). Nevertheless, NGS or second-generation technologies share their reliance on massively parallel processing and the potential to sequence thousands to millions of DNA strands simultaneously.
Next-generation technologies have undoubtedly reduced DNA sequencing costs and expedited high-quality genome sequencing. Nevertheless, Sanger sequencing remains a method of choice, especially for the analysis of low-volume DNA sequences, as it provides high-quality DNA sequencing data for regions of up to 1,000 bases.
How does Sanger sequencing work?
Sanger sequencing, or the enzymatic chain termination method, is based on the use of fluorophore-labeled dideoxynucleotides (ddNTPs) in combination with regular deoxynucleotides (dNTPs). Because ddNTPs lack a hydroxyl group needed for nucleotide binding, the addition of ddNTPS by the DNA polymerase during chain extension terminates the DNA strand elongation. Reiteration of primer annealing and DNA extension cycles results in fragments with a fluorophore-labeled nucleotide at each position, thereby identifying every nucleotide in the DNA template (Crossley et al. 2020).
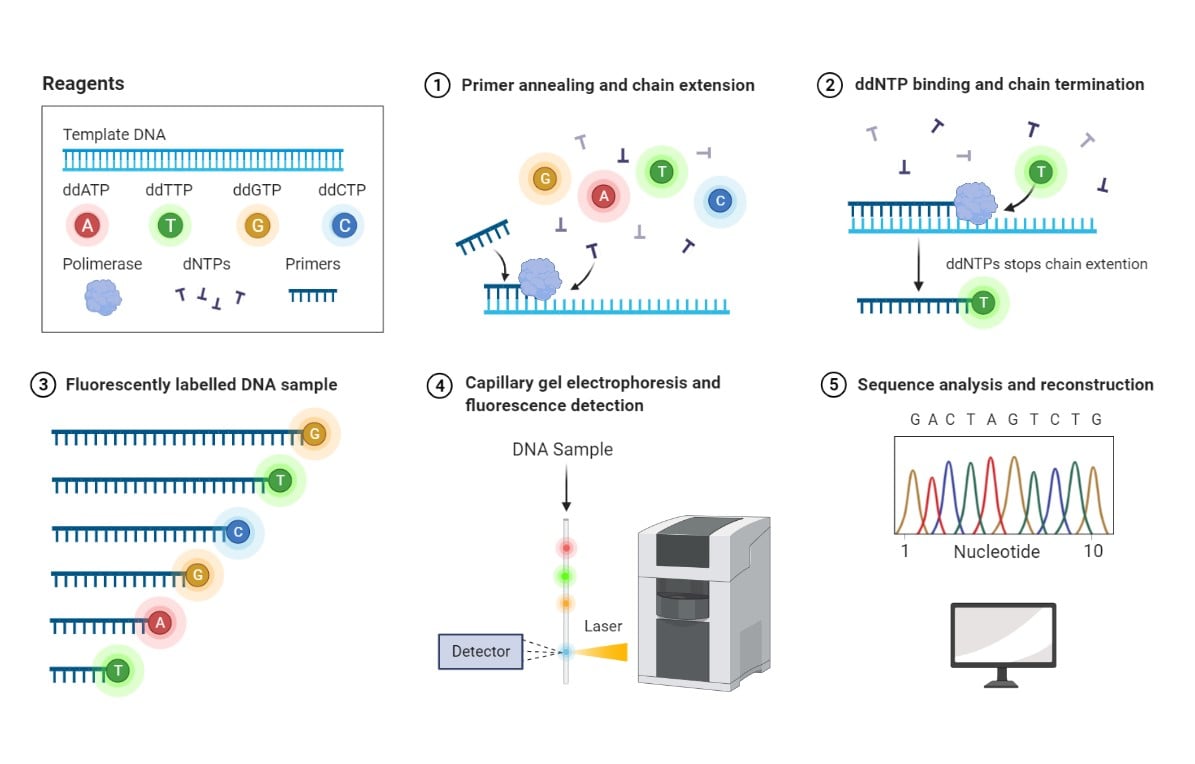
Sanger sequencing method. 1- Deoxynucleotides (dNTPs) and a low concentration of fluorophore-labeled dideoxynucleotides (ddNTPs) are supplied together with DNA template and primers. Polymerase catalyzes the DNA chain extension by random incorporation of available nucleotides. 2- Binding of a fluorophore-labeled ddNTP leads to termination of chain extension. 3- DNA fragments of various lengths and labeled with different fluorophores are generated. 4- Labeled DNA fragments are separated based on molecular mass by capillary gel electrophoresis, and their associated fluorescence is detected by a Charge-Coupled Device (CCD). 5- A chromatogram is generated based on peak fluorescence detected, corresponding to fluorophore-labeled ddNTPs incorporated at each position of the DNA sequence. Reprinted from “Sanger Sequencing”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates
Choosing between Sanger sequencing and NGS
With over 99% accuracy, the Sanger sequencing method remains the “gold standard” for basic and clinical research applications. In fact, most clinical laboratories rely on Sanger sequencing to validate gene variants (e.g., single-nucleotide variants and insertion/deletions) identified first through NGS. However, more studies are emerging supporting the accuracy of NGS methods for variant identification and questioning the redundant use of a more time-consuming and costly process for validation.
The Sanger method allows the sequencing of one DNA fragment at a time. Therefore, when it comes to choosing between next-generation and Sanger, both highly accurate DNA sequencing approaches, sample volume remains a primary differentiating factor (Slatko et al. 2018). Discovery applications requiring sequencing of a large number of genes (e.g., over 100 gene targets), or high sensitivity and therefore greater reading depth, as needed for complex samples (e.g., tumor tissue), would benefit from next-generation approaches both in cost and turn-around time.
Yet, Sanger sequencing remains a preferred method in the molecular biology laboratory for sequence verification of genes and plasmids. Partly, this is because Sanger sequencing can read up to 500-700 bps per reaction without complicated data analysis. Therefore, it is relatively easier and faster, especially for gene sequences with repeats, which remains a big challenge for NGS platforms that require linking short sequence reads together (e.g., Illumina NGS).
Pros and Cons | Sanger Sequencing | Targeted NGS |
|---|---|---|
Advantages |
|
|
Disadvantages |
|
|
GenScript routinely performs Sanger sequencing with a dideoxy ABI sequencer, a high-throughput long-read sequencing instrument, to ensure the accurate production of high-quality synthetic DNA for downstream applications. Sanger sequencing allows GenScript to guarantee a 100% sequence accuracy for the gene insert and flanking sequence, ensuring the correct reading frame. For complex, unstable sequences due to secondary structure, palindromes, and high GC content, such as the Inverted Terminal Repeats (ITR) in AAV transfer plasmids, GenScript offers a Sanger-dependent ITR specific sequencing service. The new proprietary ITR-sequencing platform enables GenScript to sequence through both ITRs, ensuring their integrity.
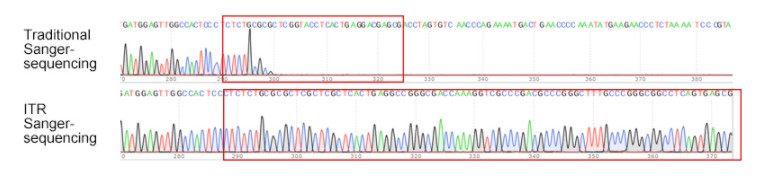
GenScript’s proprietary ITR sequencing technology. Regular Sanger sequencing typically fails to sequence through ITRs due to its high GC content, resulting in termination of the sequencing reaction and subsequent incomplete sequencing. GenScript's proprietary ITR sequencingtechnology can easily read through ITRs with clean peaks.
Analyzing Sanger sequencing results
The output of Sanger sequencing is a four-color chromatogram representing the peak fluorescence intensity associated with each labeled ddNTP along the DNA sequence. Through automated analysis, a sequence text file is also commonly generated. Nevertheless, an inspection of the chromatogram is essential for accurate sequence analysis and to resolve any potential mistakes introduced by automated base calling (Crossley et al. 2020).
Tips for Sanger sequence analysis
- Depending on the software used, sequencing chromatograms may have different formats. GenScript provides sequencing results in ab1 format. Several programs may be used to view ab1 files, such as SnapGene Viewer and FinchTV, which operate in both Mac and PC.
- Visual inspection of a high-quality DNA sequencing chromatogram should show sharp and evenly-spaced non-overlapping peaks. Overlapping peaks may indicate non-specific primer annealing or the presence of a second similar sequence.
- It is typical for sequences close to primer-binding sites to be of poor quality. Additionally, sequencing reads beyond the 700 bp range may have lower resolution depending on sample quality. Finally, far-out signals in the chromatogram (i.e., beyond 900 nucleotides) may appear as broader peaks with irregular spacing; therefore, base calling becomes unreliable.
How much do you know about DNA sequencing?
- Like (3)
- Reply
-
Share



About Us · User Accounts and Benefits · Privacy Policy · Management Center · FAQs
© 2026 MolecularCloud