IL3 in Astrocyte and Microglia Communication: A Potential Therapeutic Target


Alzheimer’s disease is a progressive neurodegenerative condition and the leading cause of dementia in the elderly worldwide. The World Health Organization (WHO) estimates that of 50 million individuals who have dementia globally, 60-70% have Alzheimer’s disease. Loss of cognitive function, such as memory and learning, gradually prevents elderly patients from independent living, increasing the burden on families and society to provide specialized support for this growing population.
What is the main cause of Alzheimer’s?
The formation and accumulation of beta-amyloid extracellular plaques and Tau neurofibrillary tangles are pathological hallmarks of Alzheimer’s disease. A direct correlation between beta-amyloid deposits and neuronal death was first proposed in the early 1990s. The “Amyloid Cascade Hypothesis” continues to be one of the prevailing drivers for therapeutic development (e.g., Biogen’s antibody-drug) to this date, and it proposes that accumulation of amyloid-beta peptides with consequent Tau neurofibrillary tangle formation is the main neurotoxicity driver (Fackhoury 2018).
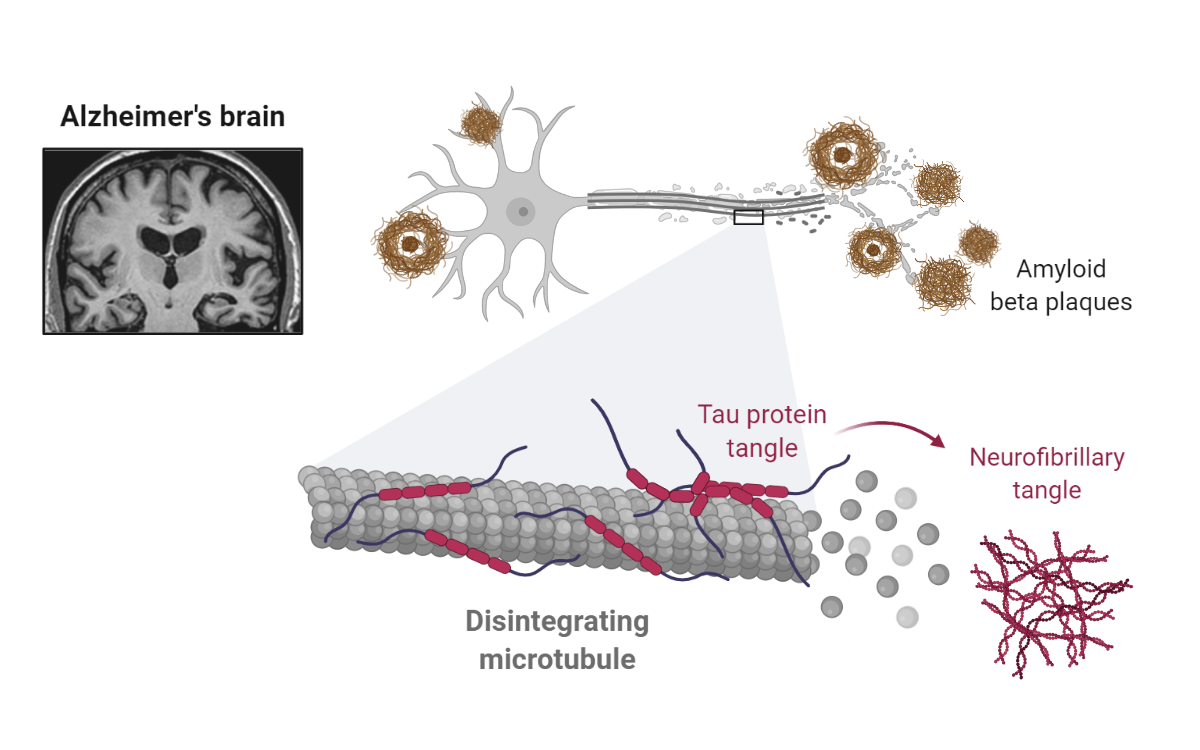
Amyloid-beta and Tau Neurofibrillary Tangles in Alzheimer’s Disease. Cleavage of the transmembrane protein Amyloid-beta precursor protein (APP) by several enzymes, such as beta-secretase and gamma-secretase, leads to amyloid-beta peptides (~40 amino acids). Extracellular accumulation of amyloid-beta peptides results in the deposition of senile plaques, which are linked to pathological Tau and neuronal cell death. Tau, a microtubule associated protein, is involved in stabilizing tubulin assembly. In Alzheimer’s disease, hyperphosphorylated Tau accumulates, forming neurofibrillary tangles in neurons. Reprinted from “Alzheimer’s Brain (Disintegrating Microtubule)”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Nevertheless, findings in Alzheimer's animal models and humans suggest that beta-amyloid deposition may not lead to neurotoxicity. Instead, evidence is accumulating for the role of other APP derivatives, such as APP’s carboxy-terminal fragments. Additionally, a link exists between Tau inclusions and Alzheimer’s disease, independent of beta-amyloid deposition (Kametani and Hasegawa 2018).
Lastly, the involvement of neuroinflammatory processes in Alzheimer’s disease, including the release of substances (e.g., cytokines) and activation of astrocytes and microglia, have more recently emerged as significant pathogenesis mechanisms (Fackhoury 2018). Although both astrocytes and microglia have been shown to participate in beta-amyloid removal, their activities can also be pro-inflammatory. Therefore, the role of glial cells in Alzheimer’s pathogenesis is far from clear as they can both promote and prevent neurotoxicity.
Astrocyte-microglia communication in Alzheimer’s disease
Recent work by McAlpine and colleagues at Harvard Medical School highlights the relevance of IL3-IL3 receptor alpha (CD123) signaling for communication between astrocytes and microglia in Alzheimer’s disease. McAlpine et al. demonstrated that in the context of Alzheimer’s disease (i.e., 5xFAD mice), IL3 expression is critical for clearing beta-amyloid and preventing cognitive decline. The team elucidated that brain-derived and not systemic IL3 expression was vital for Alzheimer’s disease protection and went on to trace the source of IL3 to a subset of astrocytes. While expression of IL3 by astrocytes was constitutive and unchanged by background (i.e., WT vs. 5xFAD), expression of the IL3 receptor was induced specifically in microglia in the Alzheimer’s brain. Significantly, McAlpine and colleagues found that IL3 receptor induction depended on TREM2 (Triggering Receptor Expressed on Myeloid cells 2) expression, a previously identified receptor critical for degradation of beta-amyloid by microglia (Zhao et al. 2018).
A deeper look at various molecular markers allowed investigators to identify a subpopulation of TREM2 positive microglia with high IL-3 receptor expression having an activated phenotype. Moreover, tampering with IL3 receptor expression in 5xFAD mice disrupted molecular programs controlling microglia morphology and motility. Indeed, overall the team found that IL3 signaling enacted molecular programs promoting microglia to navigate and cluster around beta-amyloid aggregates. Astrocyte-derived IL3 was critical to microglial activation, and its absence increased beta-amyloid deposition and reduced microglial localization to beta-amyloid aggregates.
Having found evidence of activated microglia with induced IL3 receptor expression in postmortem cortical brain samples from Alzheimer’s disease patients, investigators were confident of the relevance of this pathway in disease pathology. Lastly, McAlpine and colleagues showed that IL3, directly administered into the brain of 5xFAD, results in microglial activation and clustering around beta-amyloid aggregates and concomitant improved memory. Together, their findings provide the impetus for a therapeutic strategy targeting astrocyte-microglia communication through the modulation of IL3 signaling.

Reference
Fakhoury, M. Microglia and astrocytes in Alzheimer’s disease: implications for therapy. Curr. Neuropharmacol. (2017) doi:10.2174/1570159x15666170720095240.
Kametani, F. & Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Frontiers in Neuroscience (2018) doi:10.3389/fnins.2018.00025.
McAlpine, C. S. et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature (2021) doi:10.1038/s41586-021-03734-6.
Zhao, Y. et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron (2018) doi:10.1016/j.neuron.2018.01.031.
- Like (3)
- Reply
-
Share



About Us · User Accounts and Benefits · Privacy Policy · Management Center · FAQs
© 2026 MolecularCloud